Введение
Известно, что количество пациентов с метаболическими заболеваниями повсеместно и неуклонно растет. Ежегодно ВОЗ объявляет все большее количество заболевших сахарным диабетом (СД), что означает увеличение финансовых затрат и определяет все возрастающую нагрузку на систему здравоохранения1. Кроме того, особенностью нарушений углеводного обмена является системное поражение организма, подразумевающее развитие поздних осложнений, более высокую частоту сердечно-сосудистых событий, нередко значительные сложности в курации пациентов в пери- и послеоперационный периоды. Повышенный риск сердечно-сосудистых и цереброваскулярных событий у пациентов с СД 2 типа (СД2), несмотря на проводимую комплексную сахароснижающую, гиполипидемическую и антигипертензивную терапию, наводит на мысль о коагуляционных нарушениях в этой когорте больных с увеличением вероятности тромбообразования. Действительно, одним из механизмов повышения кардиоваскулярного риска при СД является увеличение риска тромбоза в сочетании с гипофибринолизом, что требует дальнейшего активного изучения, поскольку на сегодняшний день в мире еще не существует четких критериев для проведения антитромбоцитарной терапии у пациентов с СД или предиабетом [1–3].
Традиционно гемостаз подразделяется на первичный и вторичный. В процессе первичного гемостаза в результате повреждения эндотелия происходит вазоконстрикция, снижение образования оксида азота и простагландинов, увеличивается экспрессия фактора фон Виллебранда (фВ), его связывание с тромбоцитами и выработка активаторов тромбоцитов (аденозиндифосфат, тромбоксан А2). Результатом первичного гемостаза является образование тромбоцитарного сгустка. В результате вторичного гемостаза формируется фибриновый «каркас». Вторичный гемостаз подразделяется на общий, внутренний и внешний пути, в процессе которых активируется каскад факторов свертывания. Для восстановления сосудистой стенки и удаления тромбоцитарного сгустка включается процесс фибринолиза [4, 5].
Настоящая статья посвящена анализу изменений системы гемостаза при нарушениях углеводного обмена и их влиянию на развитие сердечно-сосудистых осложнений.
Влияние гипергликемии на систему гемостаза
Гипергликемия вызывает структурные изменения тромбоцитов, что в первую очередь нарушает состояние первичного гемостаза. Вследствие нескольких механизмов (неферментативное гликирование мембранных белков тромбоцитов, изменения свойств клеточной мембраны) происходит изменение состояния тромбоцитов в виде их гиперактивации и гиперреактивности [4, 6].
Тромбоциты у пациентов с СД2 экспрессируют большое количество маркеров активации (CD31, CD49b, CD62P и CD63), молекул адгезии (лиганд CD40) и поверхностных рецепторов (гликопротеин Ib и GPIIb/IIIa) [7]. Кроме того, существуют данные, что повышенная осмолярность при гипергликемии увеличивает экспрессию GPVI, GPIIb/IIIa и P-селектина, что приводит к усилению связи лейкоцитов с адгезивными тромбоцитами [7, 8].
Активация тромбоцитов и их участие в тромботической реакции при разрыве атеросклеротической бляшки являются важнейшими факторами, определяющими степень тромбоза, постепенный рост бляшки и развитие окклюзирующих тромбов. Повышенная адгезия тромбоцитов к стенкам сосудов, проявляющаяся ранними атеросклеротическими изменениями, и высвобождение факторов роста из α-гранул могут усугубить развитие атеросклероза. Свидетельством повышенной активности тромбоцитов у пациентов с СД является повышение концентрации в крови растворимого CD40 (высвобождаемого тромбоцитами медиатора тромбоза и воспаления) и Р-селектина (отражающего активацию тромбоцитов) [9].
Кроме того, гипергликемия через механизм неферментативного гликирования влияет на плазменное звено гемостаза, изменяя активность белков свертывающей системы крови [9].
Считается, что гипергликемия вызывает повреждение сосудов, создавая дисбаланс между биодоступностью оксида азота (NO) и накоплением активных форм кислорода, а также активных форм азота, что приводит к дисфункции эндотелия [4, 9].
Инсулинорезистентность и система гемостаза
Инсулин играет важную роль в сосудисто-тромбоцитарном гемостазе. На поверхности тромбоцитов имеются инсулиновые рецепторы, посредством которых они взаимодействуют с инсулином. В норме инсулин подавляет такие агонисты рецепторов тромбоцитов, как коллаген, приводя к снижению агрегации тромбоцитов. Инсулин также способствует высвобождению активатора плазминогена наряду с увеличением экспрессии рецепторов простациклина на поверхности тромбоцитов, что также приводит к снижению агрегации тромбоцитов [7, 9, 10]. При СД2 тромбоциты начинают проявлять резистентность к инсулину. Это, в свою очередь, снижает чувствительность тромбоцитов к NO и простациклину, молекулам, которые снижают реактивность тромбоцитов [11, 12].
В условиях инсулинорезистентности нарушается работа сигнального пути PI3K (фосфатидилинозитол-3-киназы), что приводит к снижению выработки NO эндотелиальной синтазой-NO и активации сигнального пути MAPK (митоген-активируемой протеинкиназы), что, в свою очередь, приводит к увеличению выработки эндотелина 1 и в конечном итоге к эндотелиальной дисфункции (ЭД). Инсулинорезистентность также увеличивает экспрессию ингибитора активатора плазминогена 1 (PAI-1), тканевого фактора (ТФ) свертывания крови и молекул адгезии [9, 10].
Кроме того, инсулинорезистентность стимулирует избыточное высвобождение свободных жирных кислот (СЖК) в жировой ткани, что впоследствии приводит к усилению оксидативного стресса. СЖК также при связывании с Toll-подобным рецептором усиливают выработку воспалительных факторов. К последним относят фактор некроза опухоли α и интерлейкин 6, которые усиливают экспрессию факторов адгезии в эндотелии, что еще больше усугубляет системное низкоинтенсивное воспаление, характерное для СД2. Избыток СЖК посредством ряда механизмов (выработка активных форм кислорода, нарушение выработки NO) нарушает нормальную функцию эндотелия, что было убедительно показано в серии работ, посвященных COVID-19. Таким образом, можно провести аналогию между некоторыми звеньями патогенеза и вышеперечисленными патологическими состояниями [9, 13].
Нарушения гемостаза при гипогликемии
Гипогликемия также рассматривается как один из факторов, который способствует гиперкоагуляции и повышению тромбообразования у пациентов с СД. Гипогликемия вносит свой вклад в процессы нарушения фибринолиза и активации тромбоцитов [12, 14]. При низком уровне глюкозы и высоком уровне адреналина вследствие стимуляции симпатической нервной системы наблюдается повышение активности тромбоцитов у пациентов с СД2 [15]. Вследствие воздействия воспалительных факторов и ЭД, вызванных гипогликемией, усугубляются гиперкоагуляция и нарушение фибринолиза [16]. Описано, что после эпизода гипогликемии предрасположенность к образованию тромбов сохранялась до 7 дней [17].
Дисфункция эндотелия при нарушениях углеводного обмена
Эндотелиальные клетки являются важным звеном в поддержании сосудисто-тромбоцитарного гемостаза. Эндотелий выполняет следующие функции: регуляция вазоконстрикции и вазодилатации, инициация процессов фибринолиза и тромбоза, активация тромбоцитов и управление функционированием гладкомышечных клеток [17, 18].
Известно, что особенностью ЭД при СД2 является снижение биодоступности NO, что приводит к вазоконстрикции. Это является следствием снижения в условиях инсулинорезистентности регуляции субстрата инсулинового рецептора 1 в эндотелиальных клетках [9]. Конечные продукты гликирования оказывают влияние на генерацию активных форм кислорода, что способствует экспрессии ТФ эндотелиальными клетками (а также моноцитами, макрофагами, гладкомышечными клетками). ТФ играет ключевую роль в процессе сосудисто-тромбоцитарного гемостаза. При наличии гиперинсулинемии прокоагулянтная активность ТФ вырастает на 30%, а при сопутствующей гипергликемии — на 80% [8].
Гиперкоагуляция и нарушения углеводного обмена
Отмечается связь СД 1 и 2 типов с изменениями свертываемости крови, включая патологию структуры фибринового сгустка, его формирования и лизиса. Это происходит в результате изменения концентрации и активности многочисленных коагуляционных белков [19]. Развитие хронического воспаления при СД обусловлено следующими процессами — усилением экспрессии провоспалительных цитокинов и продукции активных форм кислорода, а также активации ядерного фактора каппа В (NF-B). Известно, что хроническое воспаление приводит к активации системы комплемента и системы кинин-калликреин. Это способствует активации фактора XII, повышению концентрации прокоагулянтных факторов и белков, включая факторы VII и VIII, ТФ, протромбина и фибриногена, а также изменению активности антитромбина, усилению экспрессии тромбина [19, 20].
Для обоих типов диабета выявлено снижение концентрации естественных антикоагулянтов — протеина C и S, антитромбина III [20–22]. В свою очередь, исследование, проведенное О. Addai-Mensah et al. [23], выявило взаимосвязь концентраций протеина C и S, антитромбина III с уровнем гликемии и степенью гликемического контроля.
Нарушение фибринолиза при СД
У пациентов с СД в результате таких посттрансляционных модификаций фибрина, как гликирование и окисление, меняется структура фибринового сгустка. Выявлено, что измененный в условиях СД тромбин приводит к выработке более плотных нитей фибрина, которые являются устойчивыми к лизису сгустка [24]. Фибринолитическая система также напрямую страдает при нарушениях углеводного обмена, что еще больше способствует гипофибринолитическому состоянию. Повышенный уровень PAI-1, который является ингибитором фибринолиза, и α2-антиплазмин (α2AP), ингибирующий плазмин, были выявлены у пациентов с СД, при этом повышение включения α2AP в фибриновые сгустки отмечалось у пациентов с СД 1 типа (СД1), что повышает устойчивость к лизису фибринового сгустка. Гипергликемия приводит к гликированию плазминогена, что влияет на его превращение в плазмин и тем самым нарушает его ферментативную эффективность [19].
PAI-1 представляет собой один из ключевых ингибиторов системы фибринолиза. Повышение уровня PAI-1, которому способствует инсулинорезистентность, гипергликемия и гипогликемия приводят к снижению уровня фибринолитических факторов и в конечном счете к гипофибринолизу. Установлено, что PAI-1 повышен в 2,8 раза у пациентов с СД2 по сравнению с таковым в недиабетической группе контроля, с другой стороны, исследования показывают, что уровень PAI-1 нормализуется при компенсации СД [8, 25].
Существуют данные, что уровни других антифибринолитических белков, таких как ингибитор фибринолизиса, активируемый тромбином (TAFI), значительно выше у пациентов с СД2, а более высокие уровни TAFI коррелируют с более высоким риском тромбообразования, особенно при декомпенсированном СД [26].
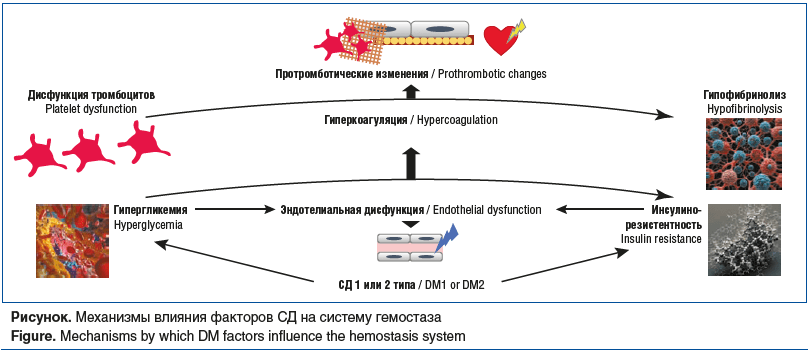
Клиническая значимость гипофибринолиза при СД2 и его вклад в увеличение кардиоваскулярного риска объективизированы в исследовании с участием пациентов с СД2 и острым коронарным синдромом, у которых более длительный лизис сгустка был ассоциирован с повышенной сердечно-сосудистой смертностью после коронарного ишемического события, несмотря на использование двойной антиагрегантной терапии [27, 28]. Схема вышеописанных патогенетических механизмов влияния СД на систему гемостаза представлена на рисунке.

Тромботический риск у пациентов с СД1 и СД2
Нарушения в системе гемостаза, выявляемые у пациентов с СД1 и СД2, имеют немало общих черт, однако необходимо отметить и значимые различия, обусловленные особенностью патогенеза заболеваний.
При обоих заболеваниях отмечается изменение концентрации ионов металлов в плазме, что приводит к нарушению регуляции коагуляции. Ионы металлов выполняют ключевые функции в системе гемостаза, регулируя функциональную активность участвующих в процессе свертывания белков. У пациентов с СД1 и СД2 наблюдается общее снижение уровня цинка [29, 30], магния [31] и повышение уровня меди [30]. При СД1 отмечается дефицит железа [32], что в конечном счете повышает риски тромбоза. У пациентов с СД2 отмечаются изменения уровня кальция, главного кофактора в каскаде активации факторов свертывания, что не обнаружено при СД1 [33]. Кроме того, у больных СД2 повышены уровни СЖК, что не характерно для пациентов с СД1. При этом повышение уровня СЖК может влиять на патологическое изменение фибринового сгустка, ЭД, образование атеросклеротических бляшек.
Уровень PAI-1 повышен при СД2, но снижен при СД1, хотя оба типа СД связаны с более длительным временем лизиса фибринового сгустка. Эти различия очень важны и должны учитываться при подборе тактики лечения. Более подробно особенности изменений в системе гемостаза в зависимости от типа СД изложены в таблице 1.
![Таблица 1. Особенности системы гемостаза в зависимости от типа СД [25] Table 1. Features of the hemostasis system depending on the DM type](https://www.rmj.ru/upload/medialibrary/f0c/h5kom5i8bjg44etc2froaok1pvo6teph/735-2.png "Таблица 1. Особенности системы гемостаза в зависимости от типа СД [25] Table 1. Features of the hemostasis system depending on the DM type")
Влияние сопутствующих метаболических заболеваний
Можно предположить, что у пациентов с ожирением и СД2 риск тромбоза выше, чем при СД2 без ожирения, поскольку в условиях инсулинорезистентности они будут характеризоваться более выраженным гипофибринолизом, более высокими концентрациями фВ и фибриногена в плазме с повышением активности факторов свертывания VII и VIII типов [28]. Кроме того, у пациентов с ожирением наблюдаются более высокие значения среднего объема тромбоцитов, уровня лептина, который ассоциирован с повышением агрегации тромбоцитов. У пациентов с дислипидемией активация тромбоцитов усиливается под воздействием фосфолипидов, содержащихся в молекулах ЛПНП. Данные окисленные фосфолипиды повышают риск тромбообразования через рецептор CD36 [34].
Влияние сахароснижающих препаратов на систему гемостаза
На сегодняшний день существует немало работ, посвященных оценке влияния сахароснижающей терапии на коагуляционный статус пациентов. Так, метформин, который является сахароснижающим препаратом первой линии и назначается чаще всего при СД2, снижает активность как фВ, так и самих тромбоцитов. Данные эффекты препарата, по-видимому, обусловлены как повышением чувствительности тканей к инсулину, так и уменьшением выраженности ЭД [35].
Препараты из группы тиазолидиндионов продемонстрировали уменьшение коагуляционных нарушений, снижая уровень фибриногена и PAI-1, тем самым минимизируя проявления гипофибринолиза, характерного для пациентов с СД [36]. Установлено, что росиглитазон дозозависимо подавлял агрегацию тромбоцитов, что было обусловлено повышением чувствительности к инсулину и снижением воспалительной реакции и выраженности оксидативного стресса [37].
Среди класса производных сульфонилмочевины, по данным исследований, глибенкламид продемонстрировал потенциальное антикоагулянтное действие, дозозависимо ингибируя экспрессию ТФ [38]. В то же время для класса препаратов данной группы характерен высокий риск развития гипогликемического состояния, что может стать причиной развития прокоагулянтного эффекта [12, 14]. Установлено, что при назначении ингибитора дипептидилпептидазы-4 (ДПП-4) вилдаглиптина достоверно снижается уровень PAI-1 [39].
Также положительное влияние на коагуляционный статус оказывают представители класса агонистов рецепторов глюкагоноподобного пептида-1 (аГПП-1), снижая низкоградиентное воспаление и способствуя экспрессии NO эндотелиоцитами и дальнейшему повышению чувствительности тромбоцитов к NO [38–40].
Ингибиторы натрий-глюкозного котранспортера 2 типа (иНГЛТ-2) тоже продемонстрировали выраженный антикоагуляционный потенциал. Так, показано, что эмпаглифлозин снижает концентрацию PAI-1 в плазме крови у пациентов с СД2 (на 25%), тем самым улучшая процесс фибринолиза [41]. Данные, касающиеся влияния сахароснижающих препаратов на систему гемостаза, представлены в таблице 2.

Применение антитромбоцитарной терапии у пациентов с СД
В настоящее время антитромбоцитарная терапия при СД в целом не отличается от таковой у пациентов с нормальным углеводным обменом. Тем не менее не вызывает сомнений, что пациенты с СД нуждаются в улучшении подходов к данной терапии.
Так, все больше доказательств того, что клопидогрел недостаточно эффективен в диабетической когорте пациентов, особенно больных СД2 [8]. По данным крупного рандомизированного плацебо-контролируемого исследования ASCEND, в котором приняли участие 15 480 человек с СД, польза от использования ацетилсалициловой кислоты (АСК) в качестве первичной профилактики сердечно-сосудистых заболеваний (ССЗ) в группе пациентов с СД нивелируется высоким процентом кровотечений (увеличение на 29% по сравнению с группой плацебо) [48].
В отечественном исследовании КАСКАД проводилась оценка эффективности АСК в буферной форме по отношению к АСК в кишечнорастворимой форме на основании данных частоты развития высокой остаточной реактивности тромбоцитов у пациентов со стабильной ишемической болезнью сердца (ИБС) и СД2. Установлено, что назначение буферной формы АСК, которая всасывается в желудке, в группе пациентов с СД2 может позволить снизить количество пациентов, не отвечающих на АСК в стандартной низкой дозе, что в дальнейшем может привести и к снижению количества значимых клинических событий без потерь в безопасности [49].
В исследовании COMPASS изучали безопасность и эффективность ингибирования тромбина на фоне применения ривароксабана (5 мг/сут) в комбинации с АСК (100 мг/сут) по сравнению с изолированным применением только ривароксабана (10 мг/сут) или АСК (100 мг/сут) у пациентов с подтвержденными атеросклеротическими ССЗ (91% пациентов с ИБС и 62% — с предшествующим инфарктом миокарда (ИМ)). Это исследование было преждевременно прекращено из-за превосходства по достижению MACE (сердечно-сосудистая смертность, ИМ или инсульт), смерти от всех причин и сердечно-сосудистой смерти в группе ривароксабана в сочетании с АСК по сравнению с одной АСК после среднего периода наблюдения в течение 23 мес. В соответствии с результатами исследования, у пациентов с СД2 (n=6922) добавление ривароксабана (5 мг/сут) к АСК приводило к достоверно более низкой частоте MACE (ОР 0,74, 95% ДИ 0,61–0,90) с более высокой частотой больших кровотечений (ОР 1,70, 95% ДИ 1,25–2,31). Столь весомые результаты продемонстрировали значение антитромбоцитарной терапии у больных СД2 и атеросклеротическими ССЗ во влиянии на сердечно-сосудистый риск, что чрезвычайно актуализирует значимость коагуляционных нарушений у подобных пациентов [50].
Для оптимизации антитромбоцитарной терапии, включая использование комбинированного лечения, необходимо проведение дальнейших исследований по изучению изменений в системе гемостаза у пациентов с различными видами нарушений углеводного обмена.
Заключение
Таким образом, нарушение углеводного обмена влияет на различные аспекты системы гемостаза. Помимо повышенной активации тромбоцитов у пациентов с СД1 и СД2 наблюдается повышение уровня и/или активности различных факторов свертывания крови в плазме, а также угнетение системы фибринолиза. Учитывая патофизиологические механизмы, лежащие в основе изменений системы гемостаза при СД, по-видимому, необходимо воздействовать как на гиперкоагуляцию, так и на повышенную активацию тромбоцитов и гипофибринолиз.
Влияние на модифицируемые факторы риска ССЗ и сопутствующие метаболические заболевания в совокупности с оптимизацией гликемического профиля позволит повысить эффективность проводимой антитромбоцитарной терапии.
Сведения об авторах:
Салухов Владимир Владимирович — д.м.н., профессор, начальник 1 кафедры терапии усовершенствования врачей им. академика Н.С. Молчанова Военно-медицинской академии; 194044, Россия, г. Санкт-Петербург, ул. Академика Лебедева, д. 6; ORCID iD 0000-0003-1851-0941
Асадова Лейла Агил кызы — аспирант 1 кафедры терапии усовершенствования врачей им. академика Н.С. Молчанова Военно-медицинской академии; 194044, Россия, г. Санкт-Петербург, ул. Академика Лебедева, д. 6.
Варавин Никита Алексеевич — к.м.н., старший ординатор кардиологического отделения 1 кафедры терапии усовершенствования врачей им. академика Н.С. Молчанова Военно-медицинской академии; 194044, Россия, г. Санкт-Петербург, ул. Академика Лебедева, д. 6; ORCID iD 0000-0001-9389-6018
Контактная информация: Асадова Лейла Агил кызы, e-mail: asadova505@yandex.ru
Прозрачность финансовой деятельности: никто из авторов не имеет финансовой заинтересованности в представленных материалах или методах.
Конфликт интересов отсутствует.
Статья поступила 28.07.2025.
Поступила после рецензирования 20.08.2025.
Принята в печать 12.09.2025.
About the authors:
Vladimir V. Salukhov — Dr. Sc. (Med.), Professor, Head of the 1 Department of Therapy for Advanced Training of Physicians named after Academician N.S. Molchanov, Military Medical Academy; 6, Akademika Lebedeva str., St. Petersburg, 194044, Russian Federation; ORCID iD 0000-0003-1851-0941
Leyla Agil kyzy Asadova — postgraduate student of the 1 Department of Therapy for Advanced Training of Physicians named after Academician N.S. Molchanov, Military Medical Academy; 6, Akademika Lebedeva str., St. Petersburg, 194044, Russian Federation.
Nikita A. Varavin — C. Sc. (Med.), Senior Resident of the Cardiology Department of the 1 Department of Therapy for Advanced Training of Physicians named after Academician N.S. Molchanov, Medical Academy; 6, Akademika Lebedeva str., St. Petersburg, 194044, Russian Federation; ORCID iD 0000-0001-9389-6018
Contact information: Leyla Agil kyzy Asadova, e-mail: asadova505@yandex.ru
Financial Disclosure: no authors have a financial or property interest in any material or method mentioned.
There is no conflict of interest.
Received 28.07.2025.
Revised 20.08.2025.
Accepted 12.09.2025.
1Всемирная организация здравоохранения. Диабет. (Электронный ресурс.) URL: https://www.who.int/ru/news-room/fact-sheets/detail/diabetes (дата обращения: 13.06.2025).