Введение
Аритмогенная кардиомиопатия (АКМП) — редкое наследственное заболевание, характеризующееся прогрессирующей фиброзно-жировой инфильтрацией миокарда и развитием жизнеугрожающих нарушений ритма и хронической сердечной недостаточности (СН) [1, 2]. Внезапная сердечная смерть (ВСС) может стать первым и единственным проявлением болезни без развития других симптомов заболевания [3, 4]. Изначально считалось, что это заболевание поражает преимущественно правый желудочек (ПЖ), что было отражено в названии патологического состояния — «аритмогенная дисплазия ПЖ» [5]. Однако в последние годы выделяют три фенотипических варианта болезни: праводоминантный, леводоминантный и бивентрикулярный [6, 7]. Два последних называют неклассическими формами АКМП. По данным литературы, левый желудочек (ЛЖ) может вовлекаться в патологический процесс в 50% случаев [8]. Следует отметить, что как диагностика данной патологии, так и определение тактики ведения пациентов с АКМП — весьма сложная задача, особенно у пациентов молодого и детского возраста, а также в случае нетипичных леводоминантных и бивентрикулярных форм. Кроме того, Критерии рабочей группы (2010) (Task Force Criteria, TFC 2010) применимы лишь в отношении АКМП ПЖ [9]. В 2020 г. для диагностики неклассических форм АКМП были предложены Падуанские критерии [10, 11]. Безусловно, развитие технологий и методов молекулярно-генетического исследования — новая веха в диагностике этого заболевания, которая позволила идентифицировать большое количество генов, ассоциированных с данной патологией, и определить генотипические и фенотипические взаимосвязи [12, 13]. Так, АКМП ПЖ наиболее часто ассоциирована с мутациями в десмосомных генах, а АКМП ЛЖ и бивентрикулярная форма — с мутациями в недесмосомных генах [7, 12]. Кроме того, известно влияние генотипа на течение и исход заболевания. Так, было показано, что пациенты с АКМП из Китая и Японии имели более высокий риск повторной госпитализации, ТС и смерти в связи с СН. Это, вероятно, было связано с различным соотношением мутаций определенных генов, таких как PKP2 и DSG2 [14].
По данным литературы, все чаще при АКМП встречается терминальная стадия СН, требующая ТС [15]. Так, S. Chen et al. [14] показали, что частота неблагоприятных исходов, включая ТС и летальный исход, вызванный СН, составляет от 2 до 22%.
Своевременное определение неблагоприятных факторов риска позволит определить персонифицированную тактику ведения пациентов и необходимость использования высокотехнологичных методов лечения (имплантация кардиовертера-дефибриллятора, ТС).
Таким образом, целью исследования стало изучение клинических особенностей и генетического спектра у детей с АКМП, перенесших ТС или находящихся в листе ожидания ТС (ЛОТС).
Материал и методы
В исследование было включено 5 пациентов (из них 3 (60%) девочки) с АКМП, перенесших ТС или находящихся в ЛОТС: 4 (80%) — с бивентрикулярной формой и 1 (20%) — с леводоминантной. Средний возраст пациентов составил 16 [7,5; 16] лет. После тщательного сбора жалоб у всех пациентов (наличие сердцебиения, синкопальных состояний, проявлений сердечной недостаточности (СН)) и семейного анамнеза выполнялись клинико-инструментальные исследования: электрокардиография (ЭКГ), суточное мониторирование ЭКГ, эхокардиография (ЭхоКГ), магнитно-резонансная томография (МРТ), эндомиокардиальная биопсия (ЭМБ) из правых камер сердца с проведением полимеразной цепной реакции ткани миокарда, молекулярно-генетическое исследование методом целевого секвенирования нового поколения с применением панели, содержащей 172 гена, наиболее часто ассоциированных с развитием кардиомиопатии. Классификация патогенности генетических вариантов была проведена в соответствии с критериями Американского колледжа медицинской генетики и геномики (2015) (American College of Medical Genetics and Genomics criteria, ACGM) [16]. По результатам исследования пациенты были стратифицированы согласно TFC 2010 и Падуанским критериям [9, 11].
Всем пациентам было назначено медикаментозное лечение (антиаритмическая терапия и терапия СН) и имплантирован кардиовертер-дефибриллятор.
Исследование было проведено в соответствии с Хельсинкской декларацией (1964) и одобрено этическим комитетом ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России (протокол от 23.01.2023 № 01-23).
Статистическая обработка данных выполнена при помощи программы Microsoft Office Excel 2019 и SPSS Statistics v.26. Учитывая редкость заболевания и небольшую численность когорты, для описания результатов исследования использовались методы описательной статистики. Показатели качественных признаков представлены в виде абсолютных значений и процентов. Учитывая количество пациентов в исследовании, для оценки нормальности распределения применялся критерий Шапиро — Уилка. Для описания количественных признаков при нормальном распределении использовали M±m, где M — среднее арифметическое, m — стандартная ошибка среднего арифметического, при ненормальном распределении — медиану с 1-м и 3-м квартилем (Ме [Q1; Q3]).
Результаты исследования
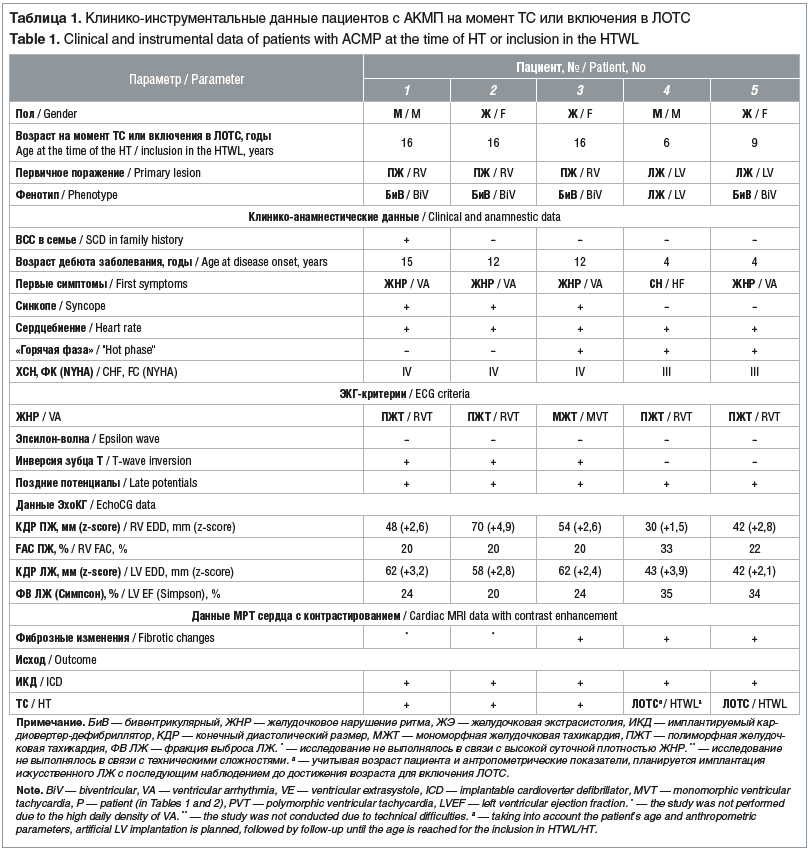
Всем пациентам согласно TFC 2010 и Падуанским критериям был установлен диагноз АКМП: 4 (80%) пациента имели бивентрикулярный фенотип, 1 (20%) — леводоминантный. Подробно клиническая картина и данные инструментальных исследований пациентов представлены в таблице 1.

Средний возраст на момент включения в ЛОТС или выполнения ТС составил 16 [7,5; 16] лет, средний возраст дебюта заболевания — 12 [4; 13,5].
Среди симптомов заболевания у всех пациентов были жалобы на сердцебиение, 3 (60%) пациента отмечали синкопальные состояния, 3 (60%) предъявляли жалобы на боли в груди. Только у 1 пациента семейный анамнез был отягощен по ВСС. Следует отметить, что у всех пациентов на момент первичного обращения в ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России уже наблюдались симптомы хронической СН, соответствующие II–III функциональному классу (ФК) согласно классификации Нью-Йоркской кардиологической ассоциации (New York Heart Association classification, NYHA). Важно подчеркнуть, что прогрессирование СН в среднем произошло в течение 3,2±1,6 года c момента дебюта заболевания. У 3 (60%) пациентов наблюдалась «горячая фаза» заболевания с эпизодами боли в груди и высокими значениями кардиоспецифических ферментов, особенно тропонина I, по данным лабораторного исследования сыворотки крови.
У всех (100%) пациентов по данным суточного мониторинга ЭКГ было зарегистрировано более 500 желудочковых экстрасистол в сутки и пароксизмы желудочковой тахикардии (ЖТ). Следует подчеркнуть, что желудочковая экстрасистолия у всех пациентов имела полиморфный характер — от 2 до 6 морфологий, в том числе с локализацией из выходного отдела ПЖ. У 4 (80%) из 5 пациентов ЖТ имела также полиморфный характер, и только у 1 (20%) ребенка была зарегистрирована мономорфная ЖТ с морфологией блокады левой ножки пучка Гиса с верхней осью.
Среди аномалий, выявленных при ЭКГ, у 3 (60%) пациентов регистрировались изменения процессов реполяризации: у 2 — инверсия зубца Т в отведениях V1–V2, у 1 — в отведениях V1–V4. Среди изменений процессов деполяризации у всех пациентов были выявлены поздние потенциалы желудочков. Эпсилон-волна не была зарегистрирована ни у одного пациента.
По данным ЭхоКГ у всех пациентов наблюдалось снижение относительно нормальных значений сократительной функции ЛЖ и ПЖ, ФВ составила 24 [22; 34,5]% и 20 [20,0; 27,5]% соответственно, выявлена дилатация ЛЖ и/или ПЖ.
Магнитно-резонансная томография сердца с контрастированием была выполнена 3 (60%) пациентам, у них были выявлены фиброзные изменения в миокарде как ПЖ, так и ЛЖ, соответствующие большому Падуанскому критерию.
Результаты ЭМБ (табл. 2), проведенной у пациентов после ТС (n=3, 60%), подтвердили морфологический диагноз АКМП. Во всех исследованных биоптатах выявлено фиброзно-жировое замещение миокарда, площадь остаточных кардиомиоцитов составила <40%, а у 1 (20%) пациента остаточные кардиомиоциты составляли всего 5–10%. Также по результатам ЭМБ у всех трех пациентов, помимо большого Падуанского критерия АКМП в виде наличия фиброзных изменений, было подтверждено наличие миокардита. При этом ПЦР эндомиокардиальной ткани не подтвердила наличие вирусного генома в кардиомиоцитах.
 results")
Результаты молекулярно-генетического исследования, выполненного всем пациентам, представлены в таблице 3. У всех пациентов были обнаружены патогенные или вероятно патогенные мутации, ассоциированные с развитием АКМП. При этом у 3 (60%) пациентов выявлены компаунд-мутации, у 1 (20%) ребенка — мутация с выпадением локуса, приводящая к потере функции белка, и еще у 1 (20%) — дигенная мутация в недесмосомных генах.

Всем пациентам была проведена имплантация кардио-вертера-дефибриллятора в целях профилактики ВСС. Троим пациентам выполнена ТС в возрасте 16 лет, и двое детей были включены в ЛОТС в возрасте 6 и 9 лет. Следует отметить, что пациенту 6 лет с учетом возраста и антропометрических показателей планируется имплантация искусственного ЛЖ с последующим наблюдением до достижения возраста для включения в ЛОТС или выполнения ТС.
Обсуждение
Аритмогенная кардиомиопатия — прогрессирующее заболевание, в ряде случаев приводящее к развитию прогрессирующей СН. Так, по данным N.A. Gilotra et al. [17], клиника СН встречается практически в 50% случаев при АКМП. Другие авторы указывают от 5 до 20% случаев развития СН при данной патологии [18]. E. Surget et al. [19] также подчеркивают частую встречаемость СН при АКМП у педиатрических пациентов, особенно с леводоминантной и бивентрикулярной формами заболевания.
В нашей работе у всех больных было выявлено вовлечение в патологический процесс ЛЖ с прогрессирующим снижением его сократительной способности и развитием клинической картины хронической СН III–IV ФК. P. Chungsomprasong et al. [20] также показали, что вовлечение ЛЖ у детей с АКМП является весьма сильным предиктором неблагоприятных исходов, включая необходимость ТС. По данным выполненного нашим авторским коллективом систематического обзора, ТС у детей с АКМП была проведена в 89% случаев при бивентрикулярной форме заболевания, в 29% случаев при АКМП ЛЖ и лишь в 0,9% случаев при ПЖ-форме патологии [21].
Следует отметить, что лишь в 1 случае манифестация заболевания была представлена СН, и, несмотря на наличие тяжелой формы желудочкового нарушения ритма, именно СН была поводом для включения в ЛОТС.
У 3 пациентов АКМП сопровождалась развитием «горячей фазы» — весьма редкого феномена. В литературе встречаются лишь единичные клинические наблюдения и исследования с небольшими выборками по изучению данного явления. Тем не менее данные литературы демонстрируют, что «горячая фаза» может встречаться при любом фенотипе АКМП, однако чаще — у пациентов с леводоминантной формой заболевания [22]. Также известно об ассоциации этого феномена с мутациями таких генов, как DSP, PKP2 и DSG2 [22], что не противоречит результатам молекулярно-генетического исследования у наших пациентов. Несмотря на то, что ряд ученых отмечает увеличение электрической нестабильности миокарда во время эпизодов «горячей фазы», что безусловно ассоциировано с увеличением риска ВСС [22, 23], тем не менее в настоящее время роль этого феномена как в прогрессировании заболевания, так и в стратификации аритмического риска неизвестна.
Следует отметить, что рядом авторов были продемонстрированы некоторые варианты мутаций в генах, ассоциированные с прогрессированием АКМП. Так, в 2015 г. А. Bhonsale et al. [24] показали, что наличие дигенных или компаунд-мутаций ассоциировано с плохим прогнозом, более ранним дебютом заболевания, высоким риском ВСС и быстрым прогрессированием СН. Это впоследствии было подтверждено в работе E. Gandjbakhch et al. в 2018 г. [25]. Ученые также показали, что риск развития СН выше при наличии мутации DSG2 по сравнению с PKP2. В то же время P.J. Scheel et al. [15] не наблюдали этого явления, что, возможно, было связано с преобладанием мутации PKP2 у больных АКМП, проживающих в Северной Америке. Известно, что у пациентов с мутациями PKP2 чаще развиваются желудочковые нарушения ритма, в то же время исследование секвенирования всего генома и секвенирования транскриптома у пациентов с пересаженным сердцем при АКМП показало, что рецессивные варианты PKP2 также могут приводить к раннему развитию прогрессирующей СН. Кроме того, на животных моделях было показано, что PKP2-укорачивающие варианты (нонсенс-мутации, сплайсинговые мутации или связанные со сдвигом рамки считывания) коррелируют с усилением тяжести прогрессирования заболевания [26]. У пациентов с мутациями гена DSP значительно чаще поражается ЛЖ [7, 8, 21]. Наличие вариантов DSP сопровождается развитием дилатационного фенотипа кардиомиопатии, дисфункции ЛЖ, СН и ВСС [7, 8].
Таким образом, столь быстрое прогрессирование СН у детей, включенных в исследование, может быть объяснено особенностями их генотипа. Так, у пациентов с мутациями в десмосомных генах имелись компаунд-мутации или мутации, связанные со сдвигом рамки считывания.
Следует отметить, что у одного пациента с подтвержденным диагнозом АКМП согласно TFC 2010 и Падуанским критериям была выявлена дигенная мутация в недесмосомных генах — патогенный вариант с известной ассоциацией с другими типами наследственных заболеваний миокарда (MYH7) и вариант с неопределенной клинической значимостью с развитием мышечной дистрофии-дистрогликанопатии (FKTN). S. Сhen et al. [14] продемонстрировали, что пациенты с одиночными и множественными мутациями могут иметь еще более высокий риск ТС или смерти вследствие СН. Кроме того, у большинства больных с множественными мутациями наблюдается по крайней мере один симптом, связанный с СН [14]. Это еще раз подчеркивает необходимость тщательного клинического и генетического обследования пациентов с последующим длительным мониторингом течения заболевания для выявления его клинических и генетических особенностей.
Нам удалось выделить ряд клинических особенностей развития заболевания, не противоречащих данным литературы: дебют заболевания в детском возрасте, поражение ЛЖ со снижением ФВ. Кроме того, тяжелое течение и быстрое прогрессирование заболевания были ассоциированы с наличием патогенных и вероятно патогенных компаунд-мутаций, мутаций с потерей функции белка, а также дигенных мутаций с вовлечением недесмосомных генов.
Стоит отметить, что в настоящее время ТС является конечной точкой в лечении пациентов с АКМП. Однако изучение и лучшее понимание генетической природы заболевания уже позволило начать разработки по созданию генной терапии для лечения больных, в частности с мутациями PKP2 [27]. Дальнейшее изучение генетических особенностей заболевания позволит ответить на многие вопросы по течению клинической картины, диагностике, прогнозу и лечению АКМП.
Ограничения исследования. У пациентов детского возраста АКМП встречается весьма редко, поэтому наша работа представлена лишь 5 пациентами, перенесшими ТС или находящимися в ЛОТС. Не всем пациентам был проведен полный объем обследования. Так, 2 детям не выполнена МРТ сердца с контрастированием в связи с высокой плотностью желудочковых нарушений ритма. ЭМБ также проведена не всем пациентам в силу их малого возраста и высокого риска осложнений данного исследования. Существенным ограничением являлось отсутствие результатов генетического обследования родителей пациентов, включенных в исследование.
Заключение
Таким образом, результаты данной работы демонстрируют прогностические инструменты, позволяющие оценить течение и исход заболевания. Дальнейшее изучение многообразного спектра генетической природы АКМП позволит решить проблему как диагностики и стратификации риска, так и лечения столь опасного заболевания.
СВЕДЕНИЯ ОБ АВТОРАХ:
Алексеева Дарья Юрьевна — к.м.н., научный сотрудник НИО неизвестных, редких и генетически обусловленных заболеваний НЦМУ «Центр персонализированной медицины» ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0003-1751-1424
Кофейникова Ольга Александровна — младший научный сотрудник НИЦ неизвестных, редких и генетически обусловленных заболеваний НЦМУ «Центр персонализированной медицины», врач детский кардиолог отделения кардиологии и медицинской реабилитации детского лечебно-реабилитационного корпуса Института перинатологии и педиатрии ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0003-4720-9023
Костарева Анна Александровна — д.м.н., директор Института молекулярной биологии и генетики, доцент кафедры внутренних болезней Института медицинского образования ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0002-9349-6257
Вершинина Татьяна Леонидовна — заведующая отделением детской кардиологии и медицинской реабилитации детского лечебно-реабилитационного корпуса Института перинатологии и педиатрии ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0003-1311-2020
Ковальчук Татьяна Сергеевна — врач детский кардио-лог отделения кардиологии и медицинской реабилитации детского лечебно-реабилитационного корпуса Института перинатологии и педиатрии ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; младший научный сотрудник НИЦ неизвестных, редких и генетически обусловленных заболеваний НЦМУ «Центр персонализированной медицины»; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0002-0842-9560
Федотов Петр Алексеевич — к.м.н., ведущий научный сотрудник НИО сердечной недостаточности, заведующий НИЛ высокотехнологичных методов лечения сердечной недостаточности Института сердца и сосудов, доцент кафедры кардиологии Института медицинского образования ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0002-7452-1971
Первунина Татьяна Михайловна — д.м.н., директор Института перинатологии и педиатрии ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России, врач-педиатр; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0001-9948-7303
Перегудина Ольга Леонидовна — врач детский кардиолог отделения кардиологии и медицинской реабилитации детского лечебно-реабилитационного корпуса
Института перинатологии и педиатрии ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0002-2761-7209
Ситникова Мария Юрьевна — д.м.н., профессор, руководитель НИО сердечной недостаточности Института сердца и сосудов, профессор кафедры факультетской терапии Института медицинского образования ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0002-0139-5177
Симоненко Мария Андреевна — научный сотрудник НИЛ кардиопульмонального тестирования НИО физио-логии кровообращения Института сердца и сосудов, врач кардиолог-трансплантолог КДЦ ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0003-3228-1188
Васичкина Елена Сергеевна — д.м.н., руководитель НИЦ неизвестных, редких и генетически обусловленных заболеваний НЦМУ «Центр персонализированной медицины» ФГБУ «НМИЦ им. В.А. Алмазова», профессор кафедры детских болезней лечебного факультета Института медицинского образования ФГБУ «НМИЦ им. В.А. Алмазова» Минздрава России; 197341, Россия, г. Санкт-Петербург, ул. Аккуратова, д. 2; ORCID iD 0000-0001-7336-4102
Контактная информация: Кофейникова Ольга Александровна, e-mail: kofeolyaa@gmail.com
Прозрачность финансовой деятельности: исследование выполнено при финансовой поддержке Министерства науки и высшего образования Российской Федерации (соглашение от 20.04.2022 № 075-15-2022-301).
Конфликт интересов отсутствует.
Статья поступила 12.09.2023.
Поступила после рецензирования 07.10.2023.
Принята в печать 30.10.2023.
ABOUT THE AUTHORS:
Darya Yu. Alekseeva — Dr. Sc. (Med.), Researcher at the Scientific Research Center for Unknown, Rare and Genetically Determined Diseases, National Center of Personalized Medicine, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0003-1751-1424
Olga A. Kofeinikova — Junior Researcher at the Scientific Research Center for Unknown, Rare and Genetically Determined Diseases, National Center of Personalized Medicine, pediatric cardiologist at the Department of Cardiology and Medical Rehabilitation of the Children’s Medical Rehabilitation Building, Institute of Perinatology and Pediatrics, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0003-4720-9023
Anna A. Kostareva — Dr. Sc. (Med.), Director of the Institute of Molecular Biology and Genetics, Associate Professor of the Department of Internal Diseases, Institute of Medical Education, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0002-9349-6257
Tatiana L. Vershinina — Head of the Department of Pediatric Cardiology and Medical Rehabilitation of the Children’s Medical and Rehabilitation Building, Institute of Perinatology
and Pediatrics, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0003-1311-2020
Tatyana S. Kovalchuk — pediatric cardiologist at the Department of Cardiology and Medical Rehabilitation of the Children’s Medical and Rehabilitation Building, Institute of Perinatology and Pediatrics, V.A. Almazov National Medical Research Center; Junior Researcher at the Research Center for Unknown, Rare and Genetically Determined Diseases, National Center of Personalized Medicine, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0002-0842-9560
Peter A. Fedotov — C. Sc. (Med.), Leading Researcher at the Heart Failure Research Institute, Head of the Research Institute of High-Tech Methods of Heart Failure Treatment, Heart and Vascular Institute, Associate Professor at the Department of Cardiology, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0002-7452-1971
Tatyana M. Pervunina — Dr. Sc. (Med.), Director of the Institute of Perinatology and Pediatrics, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0001-9948-7303
Olga L. Peregudina — pediatric cardiologist at the Department of Cardiology and Medical Rehabilitation of the Children’s Medical and Rehabilitation Building, Institute of Perinatology and Pediatrics, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0002-2761-7209
Maria Yu. Sitnikova — Dr. Sc. (Med.), Professor, Head of the Heart Failure Research Institute, Heart and Vascular Institute, Professor of the Department of Faculty Therapy, Institute of Medical Education, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0002-0139-5177
Maria A. Simonenko — Researcher at the Research Institute of Cardiopulmonary Testing, Research Institute of Circulatory Physiology, Heart and Vascular Institute, cardiologist and transplant specialist at the Consultative and Diagnostic Center, V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD 0000-0003-3228-1188
Elena S. Vasichkina — Dr. Sc. (Med.), Head of the Scientific Research Center for Unknown, Rare and Genetically Determined Diseases, National Center of Personalized Medicine, Professor of the Department of Pediatric Diseases of the Faculty of Medicine, Institute of Medical Education; V.A. Almazov National Medical Research Center; 2, Akkuratov str., St. Petersburg, 197341, Russian Federation; ORCID iD ORCID iD 0000-0001-7336-4102
Contact information: Olga A. Kofeinikova, e-mail: kofeolyaa@gmail.com
Financial Disclosure: the study was conducted with the financial support of the Ministry of Science and Higher Education of the Russian Federation (Agreement No. 075-15-2022-301 dated 04/20/2022).
There is no conflict of interest.
Received 12.09.2023.
Revised 07.10.2023.
Accepted 30.10.2023.