Введение
Врожденные нарушения обмена билирубина — это группа наследственных заболеваний, при которых происходит его накопление в крови (гипербилирубинемия), что приводит к развитию желтухи.
Существует несколько классификаций гипербилирубинемий. Наиболее распространенной является патогенетическая, которая основывается на ключевых механизмах развития патологической гипербилирубинемии. Согласно этой классификации гипербилирубинемии делятся на 4 группы в зависимости от вызвавших их причин: 1) гиперпродукция билирубина; 2) снижение клиренса билирубина; 3) повышенная реабсорбция билирубина из кишечника; 4) сочетанные нарушения [1, 2]. По происхождению выделяют наследственные и ненаследственные формы гипербилирубинемий. Наследственные формы повышения уровня билирубина, согласно патогенетической классификации, делятся по локализации дефекта в метаболизме билирубина и обусловлены патогенными вариантами (мутациями) в генах, кодирующих транспортные белки и ферменты печени. В данной статье рассматриваются наследственные формы гипербилирубинемий, связанные с нарушением клиренса билирубина.
Наследственные нарушения билирубина наследуются аутосомно-рецессивно. Наличие одного нормального аллеля обеспечивает достаточную активность соответствующего белка для поддержания нормальной функции.
По содержанию фракций билирубина наследственные гипербилирубинемии можно разделить на две группы. К первой группе относятся такие наследственные заболевания, при которых наблюдается повышение уровня неконъюгированного (свободного, непрямого) билирубина (синдром Жильбера (СЖ), синдром Криглера — Найяра I и II типов (СКН I, II)). Повышение уровня свободного билирубина связано с нарушением процесса конъюгации данного метаболита. Эта группа представляет особую опасность для новорожденных и требует повышенного внимания для своевременного установления правильного диагноза. Ко второй группе наследственных нарушений обмена билирубина относятся синдромы, сопровождающиеся повышенным уровнем конъюгированного (прямого) билирубина (синдром Ротора, синдром Дубина — Джонсона). Эта группа связана с дефектами транспорта прямого билирубина.
Целью данного обзора является подробное рассмотрение физиологической роли билирубина и систематизация современных данных о наследственных синдромах, связанных с нарушением обмена билирубина.
Метаболизм билирубина
Билирубин — это желтый пигмент, который образуется в результате распада гемоглобина и, в меньших количествах, других гемосодержащих белков. Распад гемоглобина на глобин и гем происходит в макрофагах мононуклеарной фагоцитарной системы, которые преимущественно находятся в селезенке (главный орган удаления старых эритроцитов), печени и, в меньшей степени, в костном мозге, где также разрушаются поврежденные или незрелые эритроциты [2].
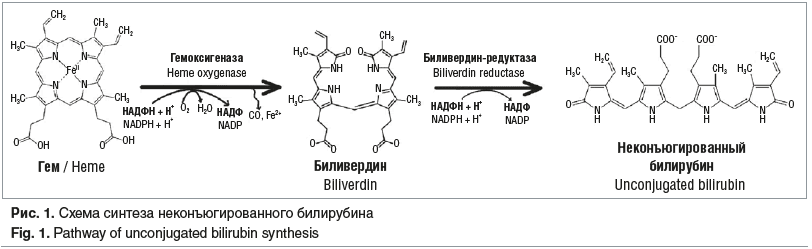
С помощью фермента гемоксигеназы гем (протопорфирин IX + Fe2+) окисляется до биливердина IXα с выделением двухвалентного железа (Fe2+) и монооксида углерода (CO). Образующийся биливердин IXα является субстратом для фермента биливердин-редуктазы, который восстанавливает биливердин до непрямого (свободного, неконъюгированного) билирубина (рис. 1). Главной химической особенностью непрямого билирубина является его неполярная (гидрофобная) структура, вследствие чего он плохо растворяется в водной среде, но легко транспортируется через фосфолипидные слои мембран, включая гематоэнцефалический барьер (ГЭБ).

Для переноса кровотоком непрямой билирубин связывается с белком-переносчиком альбумином, что обеспечивает безопасное перемещение и предотвращает его проникновение через ГЭБ. Связь между билирубином и альбумином представляет собой динамическое равновесие [3]. Под действием определенных факторов сродство между альбумином и непрямым билирубином может снижаться, в результате чего билирубин переходит в свободную (несвязанную) фракцию и способен проникать через ГЭБ, оказывая нейротоксичный эффект, особенно у новорожденных [4]. Было отмечено, что снижение связывания билирубина с альбумином наблюдается чаще у недоношенных новорожденных, чем у доношенных. Содержание свободной фракции неконъюгированного билирубина возрастает при снижении концентрации альбумина, метаболическом ацидозе, гипоксии и апноэ, конкурирующем связывании с альбумином некоторых лекарственных средств (например, сульфаметоксазола, цефтриаксона) [5].
Билирубин-альбуминовый комплекс транспортируется к печени, где происходит ключевой процесс метаболизма билирубина — его конъюгация. Перед попаданием в гепатоциты билирубин диссоциирует от альбумина.
Описаны два механизма транспорта билирубина в гепатоциты. Первым механизмом является пассивная диффузия через липидный слой мембраны — механизм flip-flop, при котором липофильная молекула билирубина способна переворачиваться в мембране и перемещаться из наружного во внутренний слой. Второй — транспорт с помощью специальных переносчиков мембранно-ассоциированных органических анионных транспортных белков OATP1B1 и OATP1B3, которые кодируются генами SLCO1B1 и SLCO1B3 соответственно [6].
В гепатоците непрямой билирубин связывается с цитоплазматическим транспортным белком лигандином, который относится к семейству глутатион-S-трансфераз. После связывания с лигандином непрямой билирубин транспортируется в гладкий эндоплазматический ретикулум, где локализован фермент уридиндифосфат-глукоронилтрансфераза 1A1 (УДФ-глюкоронилтрансфераза 1А1, UGT1A1).
UGT1A1 — фермент из класса гликозилтрансфераз, кодируемый одноименным геном UGT1A1. Данный фермент играет ключевую роль в конъюгации билирубина. Процесс конъюгации представляет собой перевод гидрофобного непрямого билирубина в водорастворимый конъюгированный билирубин посредством глюкуронидации — присоединения глюкуроновой кислоты к неконъюгированному билирубину, что обеспечивает своевременное выведение данного метаболита из организма. Данный процесс происходит в два этапа. На первом этапе образуется моноглюкуронид билирубина путем переноса одной молекулы глюкуроновой кислоты от уридиндифосфата глюкуроновой кислоты на молекулу неконъюгированного билирубина. На втором этапе присоединяется вторая молекула глюкуроновой кислоты, в результате чего образуется диглюкуронид билирубина. Продукты, полученные на этих двух этапах, являются водорастворимыми и активно транспортируются в желчь [6]. Схема метаболизма билирубина изображена на рисунке 2 (адаптирована по [7]).

После конъюгации прямой билирубин перемещается внутри гепатоцита с помощью вышеупомянутых белков лигандинов на апикальную мембрану клетки для дальнейшего транспорта. Экскреция продуктов конъюгации из гепатоцита происходит с помощью АТФ-зависимого транспортера MRP2 (мультирезистентный белок 2, белок множественной лекарственной устойчивости 2) [8].
MRP2 — это транспортный белок, кодируемый геном ABCC2, относится к семейству ABC-транспортеров, расположенных на апикальной мембране поляризованных эпителиальных клеток, его основной локализацией является каналикулярная мембрана гепатоцитов, обращенная в просвет желчных канальцев. MRP2 транспортирует моно- и диглюкурониды билирубина из гепатоцитов в желчь против градиента концентрации, гидролизуя АТФ. Если MRP2-опосредованная система перегружена, включается компенсаторный механизм обратного оттока, при котором конъюгированный билирубин поступает в синусоидальную кровь. Данный процесс опосредуется базолатеральным транспортером MRP3, расположенным на синусоидальной мембране гепатоцитов. Белок MRP3, так же как и MRP2, относится к семейству ABC-транспортеров [8], кодируется геном ABCC2. В норме MRP3-транспорт играет минимальную роль, но значительно активируется при патологических состояниях, таких как дефекты MRP2 (синдром Дубина — Джонсона (OMIM 237500)), холестаз, стресс. Цитокин фактор некроза опухоли α и киназа JNK также повышают экспрессию гена ABCC2 [9].
Повторный захват конъюгированного билирубина из синусоидального пространства в гепатоциты осуществляется вышеупомянутыми белками OATP1B1 (ген SLCO1B1) и OATP1B3 (ген SLCO1B3). С потерей функции обоих транспортеров связан синдром Ротора (OMIM 237450), который проявляется смешанной гипербилирубинемией с преобладанием конъюгированного билирубина [10]. Несмотря на то, что OATP1B1 и OATP1B3 транспортируют оба типа билирубина, особенно важна их роль в обратном захвате конъюгированного билирубина. Это связано с тем, что неконъюгированный билирубин является липофильным и способен частично перемещаться в гепатоциты с помощью пассивной диффузии, тогда как после конъюгации билирубин становится полярным и данный вид транспорта ему недоступен. Таким образом, при дефиците OATP1B1 и OATP1B3 происходит неэффективный захват прямого билирубина, что приводит к развитию гипербилирубинемии со значительной долей конъюгированного билирубина.
Косвенное влияние на транспорт конъюгированного билирубина оказывает еще одна транспортная система — BSEP (Bile Salt Export Pump), представляющая собой АТФ-зависимый насос, который активно секретирует желчные соли в желчь. Это способствует созданию осмотического градиента, обеспечивающего объемный ток желчи, под действием которого происходит пассивное перемещение прямого билирубина, уже выведенного в канальцы транспортером MRP2 [11].
Прямой билирубин вместе с желчью транспортируется по желчным протокам и желудочно-кишечному тракту, достигая толстой кишки. Здесь конъюгированный билирубин с помощью фермента β-глюкуронидазы, вырабатываемого бактериями, подвергается деконъюгации [12]. После чего высвобожденный неконъюгированный билирубин восстанавливается до уробилиногена. Большая часть уробилиногена в толстой кишке окисляется до стеркобилина и выводится с калом. Часть уробилиногена всасывается в портальное кровообращение и возвращается в печень, участвуя в энтерогепатической циркуляции. Небольшое количество попадает в системный кровоток и выводится почками, где окисляется до уробилина.
Проблема гипербилирубинемий особенно актуальна для новорожденных по нескольким причинам. В первую очередь это связано с незрелостью ферментных и транспортных систем, участвующих в метаболизме билирубина [13]. На каждом из этапов обмена билирубина сниженная функциональная активность определенных белков-транспортеров в печени может привести к появлению физиологической гипербилирубинемии и увеличить риск возникновения нейротоксических эффектов непрямого билирубина. Уровень альбумина, а также его сродство к билирубину резко снижено у новорожденных, в особенности у недоношенных, что увеличивает концентрацию свободной фракции билирубина, способной легко проникнуть через незрелый ГЭБ [5]. Экспрессия мРНК SLCO1B1 (OATP1B1) и SLCO1B3 (OATP3B3) в печени снижена, что уменьшает эффективность захвата неконъюгированного и конъюгированного билирубина. Функциональная активность фермента UGT1A1 также резко снижена и составляет менее 1% от активности у взрослых. Согласно исследованиям увеличение активности UGT1A1 до нормальных значений происходит в течение 10–20 нед. [14, 15]. Транспортер MRP2, отвечающий за экскрецию конъюгированного билирубина в желчь, функционирует, но с пониженной эффективностью [13]. Дополнительным фактором риска у новорожденных выступает недоношенность, при которой наблюдается еще большая незрелость ферментативных систем печени, высокая проницаемость ГЭБ и увеличение неблагоприятных факторов, способствующих гипербилирубинемии, которые были указаны выше [5].
Согласно клиническим рекомендациям понятие «гипербилирубинемия» в неонатологии вводится при повышении концентрации общего билирубина в сыворотке крови более 256 мкмоль/л у доношенных и недоношенных (гестационный возраст — 35 нед.), а у детей, родившихся до 35 нед. гестации, — более 171 мкмоль/л [1].
Билирубин является ключевым параметром в нескольких оценочных системах для определения прогноза и исходов заболеваний печени. Например, билирубин служит компонентом модели для оценки терминальной стадии заболевания печени (Model for End-Stage Liver Disease, MELD), параметром в шкале Чайлда — Тюркотта — Пью, предназначенной для определения тяжести циррозов печени и выживаемости пациентов, критерием в шкале ABIC, используемой для оценки тяжести течения алкогольного гепатита [7].
Помимо продукта распада гемоглобина, билирубин в нормальных концентрациях выступает в роли защитного агента для организма. Билирубин обладает антиоксидантным эффектом, обезвреживая свободные радикалы и активные формы кислорода, тем самым предотвращая перекисное окисление липидов и повреждение клеток [16]. Также билирубин обладает иммуномодулирующими свойствами, выступая ингибитором провоспалительных цитокинов и иммунных клеток [17]. Вместе с этим описано положительное влияние билирубина на липидный обмен — снижение липогенеза и стимуляция окисления жирных кислот [18].
При увеличении концентрации билирубина, особенно неконъюгированного, полезные свойства сменяются на токсические. При проникновении неконъюгированного билирубина через ГЭБ метаболит оказывает нейротоксический эффект. Билирубин нарушает функции митохондрий, вызывая нарушение энергетического обмена, а также стимулирует образование активных форм кислорода, повреждая ДНК нейронов. Эти процессы способствуют запуску апоптоза нейронов, в результате чего формируется билирубиновая энцефалопатия с переходом в ядерную желтуху [19].
Неконъюгированные гипербилирубинемии
Одной из основных причин неконъюгированных гипербилирубинемий является нарушение процесса конъюгации непрямого билирубина в печени. Основным ферментом, обеспечивающим глюкуронидацию неконъюгированного билирубина, является UGT1A1. Фермент относится к суперсемейству УДФ-глюкуронилтрансфераз (UGT), группе ферментов, играющих ключевую роль во второй фазе детоксикации ксенобиотиков и эндогенных соединений. Одним из подсемейств этого суперсемейства выделяют UGT1A, гены которого расположены в виде кластера на хромосоме 2q37.1 у человека. Генный локус UGT1A содержит до 13 первых экзонов с собственными промоторами, среди которых 4 псевдоэкзона, и несколько общих экзонов (2–5 экзоны), присутствующих у всех изоформ. Путем альтернативного сплайсинга образуется 9 функциональных изоформ, которые участвуют в глукуронидации различных соединений. В таблице 1 приведена характеристика функциональных изоформ [20].

В контексте метаболизма билирубина ключевую роль играет изоформа UGT1A1 [20]. Ген UGT1A1 содержит промоторную область, которая содержит TATA-подобный элемент в норме с повторяющейся последовательностью A(TA)6TAA. Промоторная область — участок на молекуле ДНК, расположенный перед кодирующей частью гена и содержащий сайт узнавания РНК-полимеразой. Увеличение числа повторов в промоторной области NM_000463.3:c.-53_-52insTA (A(TA)7TAA) в гомозиготном состоянии в гене UGT1A1 приводит к снижению уровня экспрессии гена и, как следствие, уменьшению функциональной активности фермента UGT1A1 [41].
Фермент UGT1A1 катализирует глюкуронидацию не только билирубина, но и ряда лекарственных препаратов, что влияет на их выведение и токсичность (иринотекан, атазанавир). Иринотекан — противоопухолевый препарат, применяемый в составе комбинированной терапии при метастатическом раке поджелудочной железы и как препарат второй линии при прогрессирующем и метастатическом колоректальном раке. Увеличение числа повторов в промоторной области в гомозиготном или гетерозиготном состояниях в сочетании с вариантами в гене UGT1A1 связано с повышенным риском развития побочных эффектов иринотекана в виде высокой вероятности возникновения нейтропении [21].
Патогенные варианты гена UGT1A1 являются причинами таких аутосомно-рецессивных заболеваний, как СЖ, СКН I, II (табл. 2).

Синдром Жильбера
Синдром Жильбера (OMIM 143500) — доброкачественное аутосомно-рецессивное заболевание, которое характеризуется умеренным повышением непрямого билирубина. СЖ является наиболее распространенным синдромом среди всех наследственных гипербилирубинемий. Распространенность его варьирует от 2 до 20% в популяции и значительно зависит от этнической принадлежности. Высокая частота синдрома, около 20%, описана у жителей Южной Азии и Ирана [42, 43]. Напротив, у жителей Восточной Азии отмечается низкая распространенность, около 2% [44]. В европейской популяции распространенность синдрома составляет от 2 до 10% в зависимости от региона [45]. Высокая распространенность заболевания выявлена в многочисленных исследованиях российской популяции и составляет около 11% [46–48].
Основным клинико-лабораторным признаком СЖ является умеренное повышение уровня непрямого билирубина в интервале 80–120 мкмоль/л с нормальным содержанием печеночных ферментов и отсутствием повреждения печени [1]. Уровень непрямого билирубина как при СЖ, так и у здоровых людей значительно варьирует в зависимости от этнической принадлежности, пола, возраста, питания и других факторов окружающей среды. Клинически синдром проявляется интермиттирующей желтухой, диспептическими расстройствами, быстрой утомляемостью. Однако в большинстве случаев заболевание протекает бессимптомно, поэтому не требует специфического лечения. К повышению уровня билирубина могут приводить такие провоцирующие факторы, как голод, физические нагрузки, инфекции, стресс, прием некоторых лекарственных препаратов [49, 50]. Также гипербилирубинемия может усугубляться у женщин в период беременности в связи с изменением гормонального статуса, что приводит к повышенной частоте и продолжительности эпизодов желтухи [51].1
В распределении по полу синдром также имеет свои особенности. Согласно данным литературы заболевание встречается чаще у мужчин, чем у женщин. Вместе с тем уровни билирубина у мужчин несколько выше, чем у женщин. Это обусловлено ингибирующим влиянием андрогенов, в частности тестостерона, на активность фермента UGT1A1 [46, 52, 53].
Одной из самых распространенных причин СЖ является увеличение числа тандемных TA-повторов в промоторной области более 6 [41]. Остаточная активность фермента у гомозигот по увеличенному числу TA-повторов составляет около 30%. Чем больше увеличено количество тандемных повторов в промоторной области, тем меньше экспрессия гена [45]. В азиатских популяциях наиболее распространенным вариантом гена UGT1A1, приводящим в гомозиготном состоянии к неконъюгированной гипербилирубинемии, является NM_000463.3:c.211G>A (NP_000454.1:p.Gly71Arg) [54].
Стоит отметить, что общий уровень билирубина превышает верхние пределы нормы не только у гомозигот по варианту c.-53_-52insTA, но и у некоторых гетерозиготных носителей [46]. Под влиянием провоцирующих факторов, таких как голод, инфекции, стресс, интенсивные физические нагрузки, уровень билирубина может выходить за верхнюю границу нормы [44, 45].
При этом у ряда гомозигот по варианту c.-53_-52insTA описано отсутствие клинической картины, характерной для СЖ. Это указывает на неполную пенетрантность данного варианта [46].
Повышенный уровень неконъюгированного билирубина оказывает негативное системное влияние на организм человека. Как упоминалось ранее, вследствие липофильности непрямого билирубина метаболит способен проникать через незрелый ГЭБ у новорожденных, что создает риск развития билирубиновой энцефалопатии. Но при СЖ этот риск остается низким по сравнению с тяжелыми формами неконъюгированной гипербилирубинемии при СКН.
Негативное влияние неконъюгированной гипербилирубинемии, связанное со снижением активности UGT1A1, проявляется в виде нарушения глюкуронидации не только билирубина, но и ксенобиотиков. Поэтому для ряда лекарственных препаратов (иринотекан, атазанавир, белиностат) существует повышенный риск возникновения побочных эффектов [21, 55].
Согласно данным исследований у пациентов с СЖ повышен риск осложнений, связанных с органами желудочно-кишечного тракта. В литературе описаны частые сочетания СЖ с заболеваниями пищевода, желудка, двенадцатиперстной кишки. Кроме того, установлено, что пациенты с данным заболеванием чаще страдают желчнокаменной болезнью [56].
Однако умеренно повышенный уровень билирубина имеет ряд преимуществ. Согласно данным клинических исследований, у пациентов с СЖ снижен риск развития сердечно-сосудистых заболеваний, атеросклеротических изменений, сахарного диабета 2 типа, метаболического синдрома, болезни Крона и колоректального рака [57–61].
У людей с данным синдромом обнаружено более медленное укорочение теломер по сравнению с контрольной группой, что дополнительно подтверждает антиоксидантное, а также потенциальное иммуномодулирующее действие билирубина [62]. Благодаря наблюдаемым антиоксидантным и провоспалительным свойствам билирубина в настоящее время активно ведутся исследования на экспериментальных моделях, направленные на изучение его терапевтического использования при различных острых и хронических состояниях [63].
Синдром Криглера — Найяра
С более выраженной неконъюгированной гипербилирубинемией связан аутосомно-рецессивный СКН (OMIM 218800, 606785), описанный в 1952 г. [64]. Синдром связан с полным отсутствием или резко сниженным уровнем фермента UGT1A1. СКН является крайне редким заболеванием [65], проявляется с одинаковой частотой у мужчин и женщин в силу тяжелого или полного дефекта фермента UGT1A1. Ярко выраженных различий в распределении между различными этническими группами не наблюдается, однако имеется высокая распространенность в изолированных популяциях вследствие повышенной частоты близкородственных браков [66].
Выделяют два типа СКН.
Синдром Криглера — Найяра I типа (OMIM 218800) характеризуется полным отсутствием фермента UGT1A1. Это обусловливает тяжелую неконъюгированную гипербилирубинемию c 15–50-кратным увеличением уровня непрямого билирубина [1]. Такой высокий уровень непрямого билирубина создает высокий риск развития билирубиновой энцефалопатии у новорожденных [67].
Причиной заболевания являются делеции, инсерции, варианты в сайтах сплайсинга, нонсенс- и миссенс-варианты нуклеотидной последовательности гена UGT1A1. Следует отметить, что патогенные варианты, расположенные в 1-м экзоне, затрагивают только изоформу UGT1A1, в то время как варианты, локализованные в общих экзонах 2–5, влияют на все изоформы, кодируемые локусом UGT1A1 [68].
Синдром проявляется обычно на 2–4-е сутки после рождения в виде нарастающей желтухи кожи и склер. При этом показатели печеночных ферментов находятся в пределах нормы, так как повреждение печени отсутствует. Главным осложнением при отсутствии лечения является развитие билирубиновой энцефалопатии и ядерной желтухи, которые обусловлены нейротоксичным действием неконъюгированного билирубина. Поражение центральной нервной системы проявляется вялостью, угнетением сосательного рефлекса, раздражительностью. На более поздних стадиях возникают такие симптомы, как судороги, апноэ, коматозное состояние, что может привести к летальному исходу. Без своевременной терапии прогноз при СКН I неблагоприятный. После развития ядерной желтухи возникшие неврологические нарушения необратимы [65].
При СКН I в качестве основного метода лечения применяется продолжительная фототерапия [69]. При достижении пороговых значений общего уровня билирубина применяется заменное переливание крови. Радикальным методом лечения СКН I служит трансплантация печени, которая, однако, имеет ряд ограничений и осложнений [70, 71]. Перспективным направлением в настоящее время является разработка генной терапии и трансплантация мультипотентных мезенхимальных стромальных клеток [72–74].
Векторной системой для генной терапии СКН выступают рекомбинантные аденоассоциированные вирусы (AAV), относящиеся к серотипу AAV8, обладающему высокой тропностью к гепатоцитам. После внутривенного введения AAV-векторы эффективно проникают через фенестрированный эндотелий синусоидов печени и трансдуцируют в гепатоциты, обеспечивая экспрессию функционального гена UGT1A1.
Результаты первого клинического исследования были опубликованы в 2023 г. L. D’Antiga et al. [73]. Согласно результатам исследования у когорты пациентов, получивших низкую дозу препарата, наблюдалось временное снижение уровня билирубина, который затем вернулся к исходному значению. У пациентов, получивших высокую дозу препарата, уровень билирубина снизился ниже 300 мкмоль/л в отсутствие фототерапии в конце наблюдения. При этом выраженных побочных эффектов не отмечалось.
В 2025 г. были опубликованы результаты двухэтапной терапии AAV8, доставляющей кодирующую последовательность UTG1A1 у ребенка с СКН I. Первая доза (6×1012 вг/кг) снизила билирубин, что позволило уменьшить продолжительность фототерапии с 12 до 4 ч/сут. Вторая инфузия в двойной дозе обеспечила дополнительное снижение уровня билирубина на 29%. Отмена фототерапии повлекла за собой резкое увеличение концентрации билирубина, в результате чего фототерапию потребовалось возобновить. Токсических побочных эффектов не наблюдалось [74].
Основным ограничением генной терапии при СКН является применение векторных конструкций у детей. Активная пролиферация гепатоцитов в раннем возрасте может привести к снижению эффективности терапии. Другим ограничивающим фактором является образование антител, направленных против вирусного капсида AAV, что также снижает терапевтический эффект препарата.
Генная терапия представляется перспективным подходом к лечению СКН и требует дальнейших исследований.
Синдром Криглера — Найяра II типа (OMIM 606785) характеризуется более мягкой клинической картиной вследствие того, что функциональная активность фермента UGT1A1 сохранена (приблизительно на 5–20%) в отличие от СКН I [65]. При СКН II наблюдается 5–20-кратное повышение содержания непрямого билирубина. Так как СКН II связан с меньшим уровнем билирубина, чем СКН I, осложнения в виде неврологических нарушений встречаются редко [1].
Большинство патогенных вариантов, вызывающих СКН II, — это миссенс-варианты, приводящие только к частичной утрате функции фермента. Высокая распространенность варианта c.-53_-52insTA в популяции может приводить к его сочетанию с патогенными миссенс-вариантами. Такой комбинированный генотип обусловливает промежуточные уровни гипербилирубинемии, что затрудняет дифференциальную диагностику между СЖ и СКН II [75].
Синдром Криглера — Найяра II типа может выявляться не только у детей, но и у взрослых [76]. СКН II может быть обнаружен при обследовании по поводу стойкой неконъюгированной гипербилирубинемии без признаков печеночной дисфункции и гемолиза. Прогноз заболевания благоприятный, из-за остаточной активности фермента UGT1A1 синдром проявляется в легкой форме или может протекать бессимптомно [65].
В качестве основного метода лечения СКН II используют фенобарбитал [1]. Снижение уровня непрямого билирубина в ответ на введение фенобарбитала является отличительным признаком, который позволяет отличить СКН II от СКН I [65]. В периоды декомпенсации может применяться фототерапия.
Несмотря на крайнюю редкость, СКН остается актуальной проблемой в связи с высокими рисками развития необратимых неврологических последствий.
Конъюгированные гипербилирубинемии
Наследственные формы гипербилирубинемий с преимущественным повышением уровня прямого билирубина связаны с дефектами транспорта конъюгированного билирубина. Заболевания с конъюгированной гипербилирубинемией протекают доброкачественно. К этой группе относятся аутосомно-рецессивные синдромы — синдром Ротора и синдром Дубина — Джонсона (табл. 3).

Синдром Ротора
Синдром Ротора (OMIM 237450) — аутосомно-рецессивное доброкачественное заболевание, проявляющееся в виде смешанной гипербилирубинемии, с преимущественным увеличением уровня конъюгированного билирубина. Данный синдром был впервые описан в 1948 г. на Филиппинах [77].
Уровень общего билирубина при синдроме Ротора составляет 34–86 мкмоль/л, в некоторых случаях больше. Конъюгированный билирубин при этом превышает 50% от общей фракции. Печеночные ферменты при данном синдроме находятся в референтных пределах. Клиническим проявлением может быть легкая желтушность кожи и склер без дополнительных симптомов [10].
Генетической причиной синдрома Ротора являются патогенные или вероятно патогенные варианты в генах SLCO1B1 и SLCO1B3, кодирующих белки-транспортеры OATP1B1 и OATP1B3 соответственно. Белки OATP1B1 и OATP1B3 (далее — OATP1B1/1B3) участвуют в обратном захвате конъюгированного билирубина гепатоцитами, а также обеспечивают альтернативный путь транспорта неконъюгированного билирубина в гепатоциты. Одновременный дефицит двух белков-транспортеров приводит к затруднению поглощения моно- и диглюкуронидов билирубина, в результате чего наблюдается повышение уровня прямого билирубина в плазме крови. Гены SLCO1B1 и SLCO1B3 располагаются рядом друг с другом в хромосомном регионе 12p12.2, для возникновения фенотипа, характерного для синдрома Ротора, необходима биаллельная (гомозиготная или компаунд-гетерозиготная) инактивация двух генов одновременно [78]. На сегодняшний день в базе данных HGMD (Human Gene Mutation Database) описано 52 варианта в гене SLCO1B1 и 30 вариантов в гене SLCO1B3.
Популяционные исследования с точной оценкой распространенности синдрома Ротора отсутствуют. По данным литературы, большинство описанных случаев данного синдрома наблюдается в Восточной Азии (Японии, Китае, Тайване) [79–81]. Патогенный вариант NM_006446.5:c.1738C>T (NP_006437.3:p.Arg580Ter) в гене SLCO1B1, приводящий к образованию преждевременного стоп-кодона, в сочетании со вставками LINE-1 в гене SLCO1B3, нарушающими нормальный сплайсинг, является распространенным вариантом у пациентов с синдромом Ротора в Восточной Азии [80]. Согласно популяционной базе данных gnomAD отдельные аллели имеют более высокие частоты у европейцев, однако для возникновения заболевания необходимо наличие биаллельных вариантов (повреждение обеих копий) в генах SLCO1B1 и SLCO1B3, как упоминалось выше [82]. В Российской Федерации описанные клинические случаи с синдромом Ротора отсутствуют.
Снижение функциональной активности белков также приводит к сниженному захвату неконъюгированного билирубина и анионных красителей, таких как бромосульфофталеин (BSP-тест с задержкой >6–12% через 45 мин), индоцианиновая зелень (ICG-тест, T1/2 5–10 мин) и 99mTc-HIDA (замедленная экскреция). По результатам сканирования HIDA (гепатобилиарная сцинтиграфия), захват печенью снижен и/или задержан, а экскреция в желчные протоки замедлена, что отличает синдром Ротора от синдрома Дубина — Джонсона [83]. Функциональные пробы позволяют подтвердить гепатоцеллюлярный дефект транспорта и установить диагноз синдрома Ротора. BSP-тест в настоящее время практически не используется в клинической практике в силу своей высокой токсичности.
Дополнительным ключевым диагностическим признаком при синдроме Ротора является повышение в моче содержания копропорфиринов [10]. Копропорфириногены (изомеры I и III) — промежуточные продукты, образующиеся при биосинтезе гема в печени и костном мозге. Часть копропорфириногенов не участвует в биосинтезе гема и окисляется до копропорфиринов с последующей экскрецией. В норме копропорфирины экскретируются: около 75% с калом (через желчь) и 25% с мочой, при этом доля копропорфирина III составляет около 80% от общего количества. При синдроме Ротора нарушена функция белков-транспортеров OATP1B1/1B3, как следствие, снижен захват органических анионов из крови в гепатоциты, в том числе копропорфиринов. В результате этого копропорфирины накапливаются в плазме крови и подвергаются почечной экскреции. Это приводит к резкому увеличению концентрации копропорфиринов в моче — в 5–10 раз выше нормы. Преимущественно увеличивается содержание фракции копропорфирина III, так как данный метаболит является основным изомером, образующимся в организме.
Также дополнительным ключевым диагностическим признаком является отсутствие патологических изменений печени, выявленных при биопсии. Данные признаки помогают дифференцировать синдром Ротора от синдрома Дубина — Джонсона [10].
Синдром Ротора является доброкачественным и не требует терапии. Прогноз заболевания благоприятный. Заболевание характеризуется многолетним течением с периодическими обострениями. Однако установление диагноза имеет важное клиническое значение. Белки-транспортеры OATP1B1/3 участвуют в захвате лекарственных веществ. Подробно описаны полиморфизмы в гене SLCO1B1, изменяющие фармакокинетику некоторых лекарственных средств. Например, rs4149056 (c.521T>C, p.Val174Ala) снижает транспортную активность белка OATP1B1. В результате уменьшается захват субстратов (статины, метотрексат, рифампицин) в гепатоциты, что приводит к повышению их концентрации в крови и увеличению риска побочных эффектов, например, при приеме симвастатина увеличивается риск развития миопатий [84].
Синдром Дубина — Джонсона
Синдром Дубина — Джонсона (OMIM 237500) — аутосомно-рецессивное наследственное заболевание, характеризующееся нарушением экскреции конъюгированного билирубина из гепатоцитов в желчь. Впервые данный синдром был описан Дубином и Джонсоном в 1954 г. [85].
Причиной данного заболевания являются патогенные и вероятно патогенные варианты гена ABCC2, расположенного на хромосоме 10q24, и кодирующего белок-транспортер MRP2. В настоящее время в базе HGMD описано более 100 вариантов, включая нонсенс- и миссенс-замены, делеции, варианты в сайтах сплайсинга.
Синдром Дубина — Джонсона считается редким заболеванием с частотой менее 1:100 000. Однако, по данным литературы, синдром часто встречается у иранских и марокканских евреев с частотой 1:1300 [86]. В Российской Федерации описанные клинические случаи данного заболевания не зарегистрированы.
Белок MRP2 является основным транспортером конъюгированного билирубина, поэтому снижение его функциональной активности может привести к нарушениям экскреции данного метаболита в желчь и, как следствие, к возникновению конъюгированной гипербилирубинемии. Общие значения билирубина составляют 50–100 мкмоль/л, в редких случаях — выше. Печеночные ферменты находятся в пределах нормы [87].
MRP2 участвует в экскреции не только билирубина, но и множества лекарственных препаратов (метотрексата, рифампицина, антибиотиков). Дефект белка-транспортера MRP2 может привести к замедлению выведения лекарственных средств и их метаболитов, что потенциально повышает риск токсических эффектов фармакологических веществ [88].
Классической особенностью синдрома Дубина — Джонсона является повышенная экскреция копропорфиринов с мочой. Но, в отличие от синдрома Ротора, преобладающей фракцией в моче является копропорфирин I. Данный метаболит образуется в основном в экстрапеченочных тканях, в том числе в костном мозге, и поступает в гепатоциты из кровяного русла. Копропорфирин III, упомянутый в разделе «Синдром Ротора», образуется в печени и костном мозге. При дефекте MRP2 копропорфирин III в большей степени образуется и задерживается внутри гепатоцитов, тогда как копропорфирин I, поступающий в печень из крови, неэффективно удерживается гепатоцитами и возвращается в системное кровообращение, что приводит к его избыточной почечной экскреции [87].
Еще одной отличительной особенностью синдрома Дубина — Джонсона является накопление темного, грубозернистого пигмента в лизосомах гепатоцитов, из-за чего в некоторых случаях печень может выглядеть черной [89].
Также для диагностики данного заболевания используется холесцинтиграфия, которая показывает характерную картину отсутствия визуализации желчного пузыря и желчных протоков в присутствии интенсивной, однородной и длительной визуализации печени [90].
Большинство пациентов с синдромом Дубина — Джонсона не имеют симптомов, за исключением желтухи. Как правило, заболевание встречается равновероятно у мужчин и женщин и проявляется в подростковом или молодом возрасте. Однако описаны случаи манифестации заболевания в неонатальном периоде, при которой в редких случаях возникали такие симптомы, как холестаз и гепатомегалия [91, 92].
Синдром Дубина — Джонсона является доброкачественным и не требует специфического этиотропного лечения. Прогноз при заболевании часто благоприятный, продолжительность жизни не изменяется. В качестве симптоматической терапии у новорожденных при тяжелых состояниях показано применение фенобарбитала и дезоксихолевой кислоты [87].
Заключение
Таким образом, рассмотренные в данном обзоре наследственные формы гипербилирубинемий (СЖ, синдром Дубина — Джонсона, синдром Ротора) характеризуются доброкачественным течением и не требуют специфического лечения. Исключением является СКН, в частности I типа. Заболевание представляет особую опасность для новорожденных и может привести к тяжелым неврологическим последствиям.
Помимо рассмотренных форм гипербилирубинемий существуют другие наследственные формы, которые характеризуются опосредованным повышением уровня билирубина, а также патологические состояния, связанные с повреждением печени или нарушением оттока желчи. В этих случаях дифференциальная диагностика помогает вовремя установить причину гипербилирубинемии и своевременно назначить правильную терапию.
Важно отметить, что все синдромы, рассмотренные в данной статье, связаны с нарушением функциональной активности белков-транспортеров и ферментов. Указанные белки являются переносчиками не только билирубина, но и ряда ксенобиотиков. Дефицит этих ферментов может привести к изменению фармакокинетики некоторых лекарственных средств, что стоит учитывать при назначении терапии пациентам с патогенными вариантами в соответствующих генах.
Сведения об авторах:
Лендоева Дарина Викторовна — младший научный сотрудник лаборатории ДНК-диагностики, врач — лабораторный генетик лаборатории молекулярно-генетической диагностики 1 ФГБНУ «МГНЦ»; 115478, Россия, г. Москва, ул. Москворечье, д. 1; ORCID iD 0000-0002-4024-8540
Семенова Наталия Александровна — к.м.н., ведущий научный сотрудник научно-консультативного отдела, врач-генетик, доцент кафедры медицинской генетики ФГБНУ «МГНЦ»; 115478, Россия, г. Москва, ул. Москворечье, д. 1; ORCID iD 0000-0001-7041-045X
Дегтярева Анна Владимировна — д.м.н., профессор, заведующая отделом педиатрии института неонатологии и педиатрии ФГБУ «НМИЦ АГП им. В.И. Кулакова» Минздрава России; 117997, Россия, г. Москва, ул. Академика Опарина, д. 4; профессор кафедры неонатологии ФГАОУ ВО Первый МГМУ им. И.М. Сеченова Минздрава России (Сеченовский Университет); 119991, Россия, г. Москва, ул. Трубецкая, д. 8, стр. 2; ORCID iD 0000-0003-0822-751X
Щагина Ольга Анатольевна — д.м.н., первый заместитель директора, доцент, заведующая кафедрой молекулярной генетики и биоинформатики, врач — лабораторный генетик лаборатории молекулярно-генетической диагностики ФГБНУ «МГНЦ»; 115478, Россия, г. Москва, ул. Москворечье, д. 1; ORCID iD 0000-0003-4905-1303
Поляков Александр Владимирович — д.б.н., профессор, член-корреспондент РАН, заведующий лабораторией ДНК-диагностики, главный научный сотрудник лаборатории ДНК-диагностики, профессор кафедры молекулярной генетики и биоинформатики ФГБНУ «МГНЦ»; 115478, Россия, г. Москва, ул. Москворечье, д. 1; ORCID iD 0000-0002-0105-1833
Прозрачность финансовой деятельности: никто из авторов не имеет финансовой заинтересованности в представленных материалах или методах.
Конфликт интересов отсутствует.
Статья поступила 19.03.2026.
Поступила после рецензирования 13.04.2026.
Принята в печать 30.04.2026.
About the authors:
Darina V. Lendoeva — Junior Researcher at the Laboratory of DNA Diagnostics, Laboratory Geneticist at the Laboratory of Molecular Genetic Diagnostics 1, Research Centre for Medical Genetics; 1, Moskvorechye str., Moscow, 115478, Russian Federation; ORCID iD 0000-0002-4024-8540
Nataliya A. Semenova — C. Sc. (Med.), Leading Researcher at the Consultative Department, Geneticist, Associate Professor at the Department of Medical Genetics, Research Centre for Medical Genetics; 1, Moskvorechye str., Moscow, 115478, Russian Federation; ORCID iD 0000-0001-7041-045X
Anna V. Degtyareva — Dr. Sc. (Med.), Professor, Head of the Pediatrics Department at the Institute of Neonatology and Pediatrics, V.I. Kulakov National Medical Research Center for Obstetrics, Gynecology and Perinatology; 4, Akademika Oparina str., Moscow, 117997, Russian Federation; Professor at the Department of Neonatology, Sechenov First Moscow State Medical University; 8 Build. 2, Trubetskaya str., Moscow, 119991, Russian Federation; ORCID iD 0000-0003-0822-751X
Olga A. Shchagina — Dr. Sc. (Med.), First Deputy Director, Associate Professor, Head of the Department of Molecular Genetics and Bioinformatics, Laboratory Geneticist at the Laboratory of Molecular Genetic Diagnostics 1, Research Centre for Medical Genetics; 1, Moskvorechye str., Moscow, 115478, Russian Federation; ORCID iD 0000-0003-4905-1303
Alexander V. Polyakov — Dr. Sc. (Bio.), Professor, Corresponding Member of the Russian Academy of Sciences, Head of the Laboratory of DNA Diagnostics, Chief Medical Researcher, Professor at the Department of Molecular Genetics and Bioinformatics, Research Centre for Medical Genetics; 1, Moskvorechye str., Moscow, 115478, Russian Federation; ORCID iD 0000-0002-0105-1833
Financial Disclosure: no authors have a financial or property interest in any material or method mentioned.
There is no conflict of interest.
Received 19.03.2026.
Revised 13.04.2026.
Accepted 30.04.2026.
1The Genotype Tissue Expression (GTEx) Project [Internet]. (Electronic resource.) URL: https://gtexportal.org (accesse date: 23.01.2026).